
 1 BBSP – Building Blocks Structure Predictor
1 BBSP – Building Blocks Structure Predictor
Secondary structures identified by HCAM method, as well as both vdW locks and closed loops collected in the
PBBD, may provide fingerprinted maps for the identification of several building blocks in both
prokaryotic and eukaryotic proteins. Thus, we linked HCAM profiles to an own made hybrid
combinatorial fragment assembly algorithm that is based on dynamic programming and that leads to
prediction of protein three-dimensional structures through generation of hypothetical assembled
models, followed by structural comparisons between such models and database building blocks. The
template-based algorithm described here is named Building Blocks Structure Predictor (BBSP)
(see Figure 1).
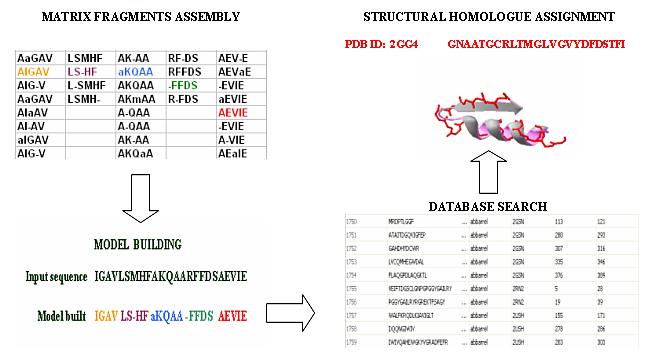
Figure 1. Schematic representation of BBSP algorithm. First, fragments stored in the BBSP matrix are assessed and assembled recursively, in order to build hypothetical structural models. Finally, assembled structural models are compared with members of the PBBD and refined by secondary structure and physicalchemical profile search, in order to calculate scores, hence retrieving the best structural matches from the database.
1.2 BBSP Results
Predictive tests on an independent protein set varying in length from 90 to 250 aa demonstrated
the capacity of BBSP to identify folding motifs with a high grade of structural homology, though with
weak sequence identity (usually < 20%). Overlapping of structural models built by the BBSP assembly
algorithm showed that such method can provide structural information on the type of folding
identified, since backbone RMSD values of building blocks belonging to the same structural category
and having similar lengths often fall within the crystallographic range. In many cases, motifs
belonging to the unclassified/disordered sub-set also revealed a high grade of overlap with target
native models. Secondary structure prediction rate of matched models refined by using HCAM score
was estimated at about 82%. Nevertheless, hydropathy clustering assistance allowed the detection of
super-secondary models having RMSD closer to target resolution (see sample results downloadable at
Tools section). For each set of results obtained, it was almost always possible to find at least one
structural model having RMSD below 1.5 Å. Furthermore, 74% matched fragments fall within the PDB
range resolution; whereas the mean RMSD of the whole population was estimated at 2.3 Å. Moreover, in
each generated sub-set, it was often possible to detect at least one full length predictive model
that covers target sequences corresponding to a super-secondary motif, though for building blocks
over 45 amino acids there is a low probability to find a full size assembled counterpart. In many
cases, it is worthy to note that when main chain trajectories of several predictive segments are
joined together, it was possible to obtain accurate prediction of higher order architectures. The
secondary structure profiles generated by HCAM and used to aid the alignment procedure often made it
possible to more correctly identify protein fragments having backbone trajectories that replicate
target super-secondary folds. Our matches were compared with the ones detected by HHsearch/HHpred,
which is one of most cited protein remote homologues detection methods in the scientific literature.
The search was conducted on the standard PDB database, using 8 PSI-BLAST iterations and local
alignment mode (HHpred web server at: http://toolkit.tuebingen.mpg.de/hhpred).
The average number of predictive matches found by HHpres was, as expected, higher than that found by our method: HHpred
can perform an extensive search on the whole updated PDB database by
position-specific scoring
matrices (PSSMs), while BBSP performs search through our in-house database only. Average RMSD of
models detected by HHpred reveals a better accuracy than BBSpred ones. However, both methods
detected backbone trajectories enclosed within the PDB resolution of native targets. On the contrary,
BBSpred was able in many cases to detect remote homologues having sequence identity significantly
lower than HHpred counterparts (see Table 1). Therefore, we argued that our method can improve
sensitivity in the detection of remotely related sequences, owing to the building of hypothetical
models by mix and joining different sized fragments that are excised from unrelated structures. Instead,
HHpred performs a multiple sequence alignment of related protein sequences stored in public databases,
with no mixing of detected models. Hence, our search strategy might have extended coverage of
conformational space required for the detection of almost sequence-independent related structures.

Table 1. Secondary structure prediction rate (Q3),
Sequence identity (Seq. id.) and backbone Root Mean Square Deviation (RMSD) for super-secondary
structure models detected by both BBSP and HHpred methods. Sequence positional numbers are
enclosed within brackets. Symbol * is referred on HHpred results having gaps in matched sequences.
1.3 Detection of protein domain superfamilies/superfolds
For the purposes of our work, the only detection of short motifs would have been reductive.
Accordingly, we tested our method on five whole protein sequences in order to detect domain
superfamilies/superfolds by the merging of trajectories of contiguous motifs (see Figure 2). Our results were compared with the ones obtained by the protein fold-recognition servers HHPred, COMPASS, and PHYRE2 .
Proteins used in our test set were represented by Streptococcus agalactiae hyaluronate lyase (PDB ID: 1F1S), Pseudomonas putida muconate lactonizing enzyme (PDB ID: 1BKH), Acinetobacter
glutaminasificans glutaminase-asparaginase (PDB ID: 1AGX), Salmonella typhimurium N-acetylgamma-glutamyl-phosphate reductase (PDB ID: 2G17), and Lactococcus lactis 6-phosphogluconate dehydrogenase (PDB ID: 2IZ0). Despite the reduced size of our library, BBSP was able to detect six folds sharing significant remote homology with respect of the targets. Particularly, both Enolase C-terminal domain of Pseudomonas putida (D)-glucarate dehydratase (PDB ID: 1BQG) and NAD (P)-binding Rossmann-fold of Sinorhizobium morelense 1, 5-anhydrod-fructose reductase (PDB ID: 2GLX), revealed the lowest sequence identity. Moreover, GaGLyase fold of Streptococcus pneumoniae hyaluronate lyase (PDB ID: 1EGU) shown the best
accuracy in terms of both RMSD and TM-score. Furthermore, BBSP was able to detect two
architectures corresponding to spatially contiguous folds of 1AGX protein, represented by one
NAD(P)-binding Rossmann fold of Escherichia coli Succinyl-CoA synthetase (PDB ID: 1CQI), and
one L-asparaginase-like superfamily domain of Escherichia coli L-asparaginase (PDB ID: 3ECA).
In four cases, COMPASS was not able to detect templates, whereas HHPred outperformed in two
cases by the detection of remote homology domains, as well as PHYRE2. Overall, BBSP results
resembled those obtained by the other methods. Finally, the search strategy of BBSP algorithm
demonstrated that domain-building motifs of our library could be successfully used as a probe to
detect higher order protein architectures.
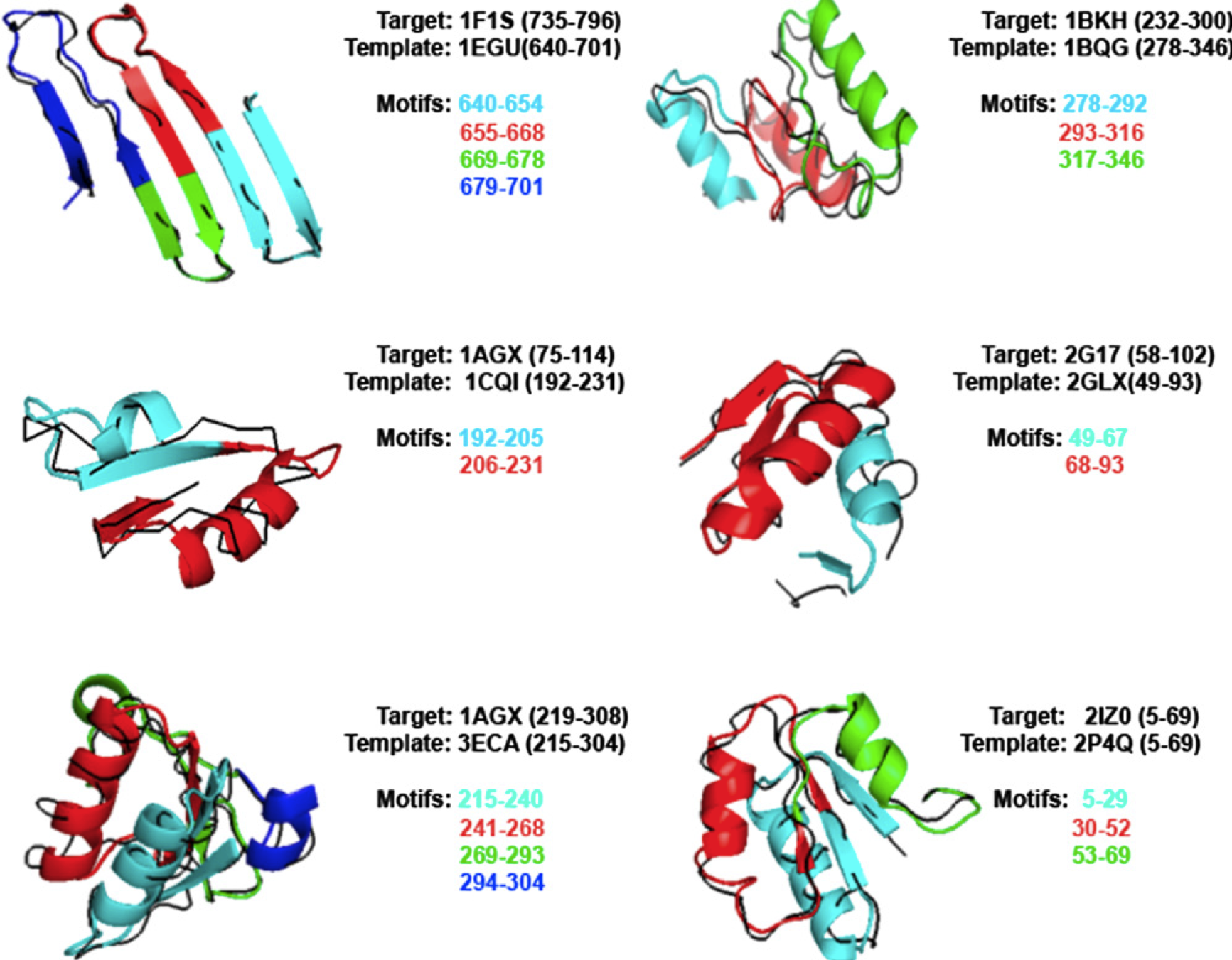
Figure 2. Domain superfamilies/superfolds detected by BBSP algorithm by concatenation of trajectories of domain building motifs. Domains are represented as black ribbon, whereas motifs are represented as color-coded Q3 cartoon.
1.4 Conclusions
Despite the time consuming calculation needed, the method discussed in this section has
been completely automated and does not require expensive computational resources. Moreover,
BBSP can be easily parameterized to run parallel execution and to define matching parameters (read instructions contained in the BBSP zip file downloadable from the Tools section). The secondary structure prediction module takes only few seconds for generating the results, whereas it
was estimated that BBSP requires from 3 to 8 hours of parallel execution (depending from the
complexity of the dynamic matrix) for a 150 mers sequence if tested by Intel® Core Duo 3.2 GHz
CPU and 4 GB RAM. Structural results were obtained solely by rigid transformation protocols and could provide guidelines for the design of both globular and transmembrane proteins. Indeed, BBSP can generate its own models of great structural relevance and can therefore be a valid starting point for the development of molecular statistical mechanics protocols. Furthermore, the predictive skills of such method will be refined by adding new statistics. Nonetheless, filters to detect false positive matches will be built. In short, BBSP could be considered as a remarkable complementary tool in the field of fold-recognition.
Reference
· Gullotto D, Nolassi SM, Bernini A, Spiga O, Niccolai N. Probing the protein space for extending the detection of weak homology folds. J Theor Biol 320, 152-158 (2013).